AI Agents should curate biological data
But getting them to be reliable is hard!

Here is a use case for an AI Agent that would make life easier for CompBio researchers:
Download a single-cell omics dataset, and accurately extract harmonized sample-level metadata.
Given such an agent, you could imagine letting it loose on all published datasets, giving you a hassle-free and ever-expanding corpus of data. This corpus can in turn form the training data for the first “virtual cells.”1
There has been a lot of activity in this area recently: the Arc Institute released SRAgent and the associated scBaseCount data corpus. The SRAgent is quite impressive: it can peek at the raw data and infer the correct CellRanger pipeline to run, and it was able to process hundreds of millions of cells this way. However, it can currently only collect a few bare-minimum metadata fields (and is missing some important ones like donor_id), so there is clearly room for a specialized curation agent.
As faith would have it, I saw three (!) separate posts about this last week: LatchBio released an agentic framework, Elucidata released a multi-agent system, and Tommy Tang used an AI worker to curate a dataset,
I have had to wrangle my fair share of datasets over the years, I have worked with most of the data vendors2, and I have played a lot with AI agents, so I have some thoughts to share. First, I’ll explain the metadata curation problem from the human perspective, then I’ll speculate on the challenges of translating these steps into an agentic system. The big challenge, of course, is reliability and accuracy (see also “Can you trust AI to clean your data?”).
I believe that metadata curation is the kind of problem where one can put together an impressive-looking demo over a weekend, but getting to several nines of reliability is really, really hard.
How these agents can be implemented to be as accurate as possible remains an open question, but what’s certain is that the field would benefit from step-by-step benchmarks.
The problem
Imagine you’re a computational biologist working on bone marrow disorders, and you see that a new single-cell paper on aplastic anemia was just published. These studies are rare, so you’re super excited to dive into the data. Can you reproduce what the authors found? Do you see any aberrant cell states? How does the data compare to previous studies?
However, the excitement only lasts so long. You sigh deeply. Now comes the slog: downloading the data from GEO, formatting it right, searching through the supplementary files. If you’re lucky, the authors give you a clean Excel file with some donor information. If you’re less lucky, the authors hand you a table spread across multiple pages of a PDF. You grit your teeth and spend two hours cleaning up the data, and now the real work can start.
You think to yourself: in a world where the latest AI models can do well in advanced math benchmarks, surely they can help you clean your data? You imagine how nice it would be: just tell the AI agent what fields you want, and it dutifully starts inspecting the paper, the supplementary files and the SRA database, and within minutes spits out a table. You look at the result, and you couldn’t have done it better yourself! If only you could get rid of this massive activation energy to get started, you could build on the momentum from that initial excitement when you first saw the paper.3
The task
Metadata curation has a few key steps:
Extracting metadata for each biological sample: use your schema - a set of fields that you want to extract: donor, age, sex, tissue, disease, sequencing technology, etc.
Harmonizing: using consistent terms for each answer.
As a data scientist, you are happy when a “B cell” is always called a “B cell”, and not “B cells” or “b cell”. This lets you do queries across many datasets.
You would typically use an ontology for this4. You can think of an ontology as a tree of pre-curated terms and their parent-child relationship. For example, if you choose aplastic anemia from the MONDO ontology , you know from the tree structure that it is a subset of anemia, which is a subset of hematologic disorder, which is a subset of human disease.
Extracting “everything else”: each dataset will surely have useful donor-level information that you didn’t capture in your schema - you can’t possibly have a standardized vocab for everything under the sun. You still want to include this information as-is into your curated object. Note: compendia like CellXGene lack this!
Now that we know what to do, let’s see how a human would think through the problem.
The human perspective
The self-talk starts here.5
Let’s start by glancing at the paper I just mentioned:

We see that the data is available in GEO. Let’s take a look at the accession:

Already, we can breathe a small sigh of relief – it’s a nice dataset.
A nice dataset satisfies three criteria:
the data lives on SRA/GEO (the 800-pound gorilla of sequencing data)
it has a link to a publication
said publication is freely accessible on PubMed Central
We’ll talk more later about how to deal with non-nice data.
It looks like there are 182 samples from multiple donors. Some have a “3M” sample, others a “6M” sample, or a “GEM baseline”. We’d have to learn more about the exact study design to understand what these abbreviations mean. Let’s click on the first sample:
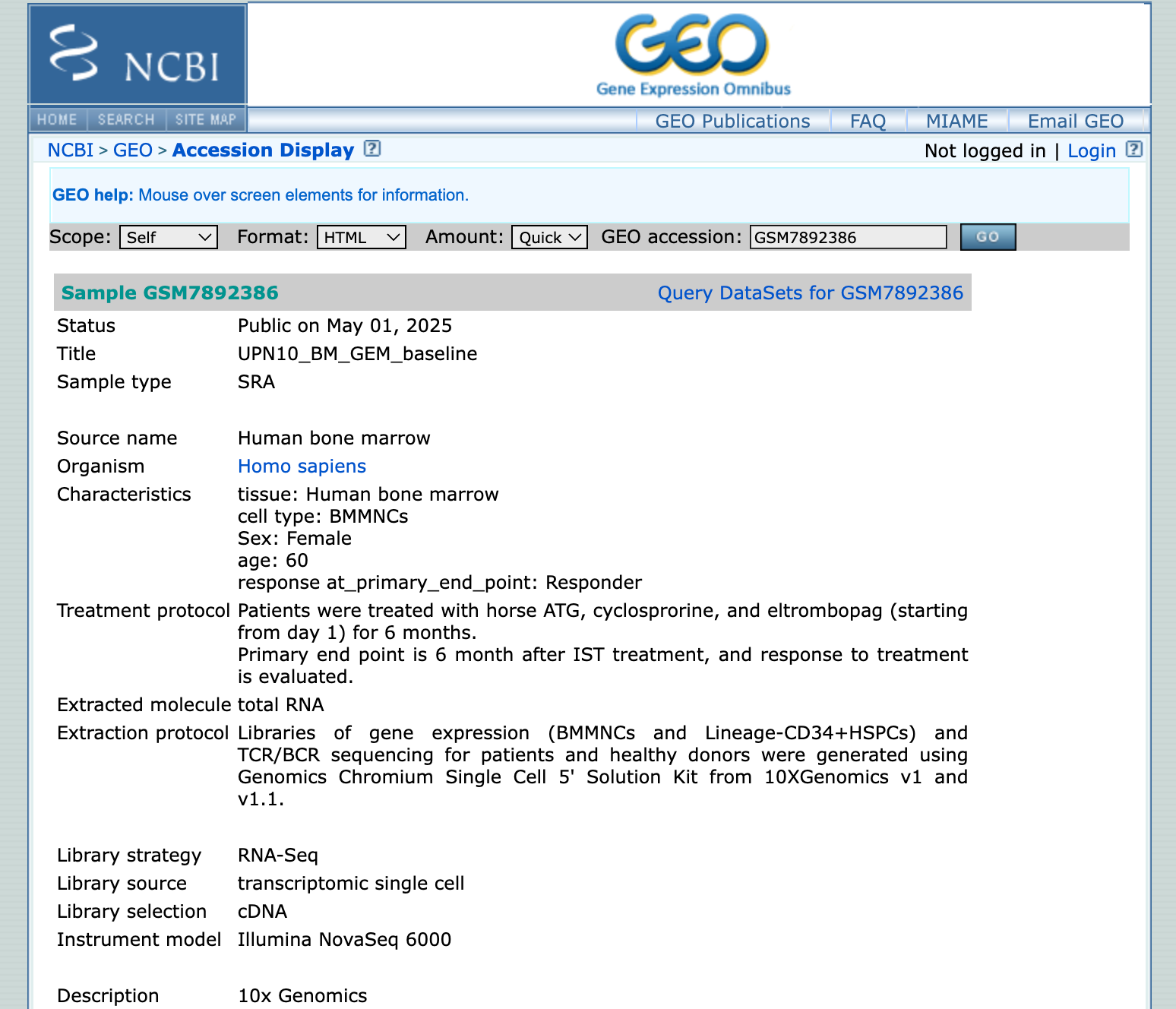
So these are bone marrow mononuclear cells from a 60 year old woman that responded to treatment. We see the treatment protocol, the 10x chemistry, and we see that they also did TCR/BCR sequencing. This is a great start!
We wonder if there is more information in the supplemental data. This is the first Excel file we download:
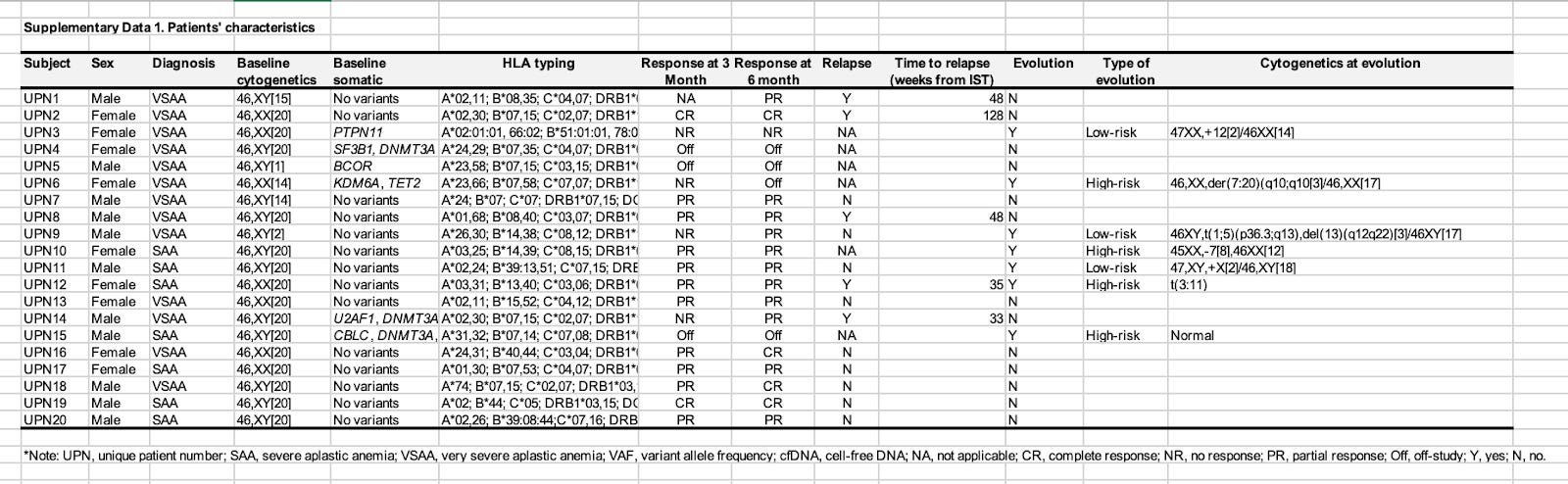
What a treasure trove of information! We get the exact diagnosis, a detailed response status, cytogenetics and HLA typing.6
Now comes what the Elucidata preprint calls sample-donor mapping.
As a scientist, you can often just look at the GEO page and the Excel file side by side, squint, and it typically becomes clear how the information relates.
In this case, we’re lucky that it’s easy!
UPN10 is a donor ID, and we see many samples on GEO where the name starts with UPN10, so obviously those belong together. We join the two tables together.
Now that the data is in the right shape, we do the work: for each sample, we annotate the gender, age, disease, treatment, response status. We check that the answer matches an ontology term. And then we make sure to keep all other columns from the Excel file above. This process usually takes at least an hour, but often more.7
Surely, AI could do this job! But how do we get it reliable enough that we can trust it?
Each curation step needs its own evals
Let’s revisit the steps above and consider the challenges when translating this into an AI system.
Finding the paper
In our example above, the GEO page has a link to the paper, but this is often missing. You’d need an agent that can search Google, inspect the search results, and identify when a paper actually belongs to a dataset or not.8
Getting the data9
We mentioned that the example dataset above is nice. However, I estimate that this is the case for only ~70% of all public single-cell data.10 The core challenge is clear: even if your AI can annotate each field perfectly, it still needs access to all the input files, and there is a long tail of non-nice data for which this creates schlep.11
Parsing the supplementary files
To the human eye, the table we saw earlier was pretty neat. Still, machine processing comes with complexity: merged cells that need to be unmerged, tables split across multiple pages, a legend below the table (or even separately in the paper). To make things worse, tables might be in PDF form! Elucidata uses GPT 4.1’s vision capability for this. From my experience, vision models work perfectly in clear-cut cases (and are surely better than old-school OCR), but they still make the catastrophic mistakes occasionally.12
Sample-donor mapping
As we established above, here the LLM basically needs to do a table join between the table on GEO and the Excel file: for each sample on GEO, what is the corresponding donor row in the spreadsheet? It’s like a table join, but you don’t have a stable ID to join on.
Extraction and harmonization
Here, the agent needs to jointly consider information from the paper, the GEO, and the table. For example, if the paper says that there were follow-up samples at 3 and 6 months, that might help the LLM understand “3M” and “6M” in GEO. The context window used to be an issue here, but no more - these days, you can stuff all information in the prompt!
What this emerging field desperately needs is a benchmark with evals for each of these five sub-tasks. Whenever the agent makes a mistake, you want to be able to pinpoint the root cause. Steps 3 and 4 are the least glamorous, but a mistake in those can be catastrophic down the line. Conversely, assuming you correctly do steps 3 and 4, I’m confident that today’s LLMs can already nail step 5 as least as well as humans can.
Routers versus autonomous agents
We’ve established that accuracy is the name of the game. If we’re going to outsource this task to an agent, we need to trust it! How do we get there, especially when LLMs can hallucinate? How do we implement guardrails so that the agent doesn’t go off-track?
The main tradeoff here is reliability versus control.

Graph taken from LangChain Academy
We could choose to build a router, which looks a lot like regular ol’ software. It only inserts an LLM when necessary, in carefully chosen places, and always padded with “bubble wrap” to ensure misbehavior is handled.13 “Think with the model, but plan with code.” In our example, we’d need to explicitly translate the step-by-step human thought process into code. We lay out the recipe, and at each step, we’d carefully manage the information the model gets to see in the prompt.
This requires a lot of engineering!
Contrast all of this to the end-to-end autonomous agent, which has free reign. It simply absorbs all of the documents, and relies on its general reasoning capability to answer the question. Scale-pilled proponents will draw the analogy to the linguists and the bitter lesson: they’ll say that more test-time compute will ultimately beat expert-constructed recipes, just like more training data ultimately beat clever handcrafted features.
It’s interesting to see where current approaches fall on the router-versus-agent spectrum. Elucidata takes the middle ground: they split the problem into multiple subtasks, with each subtask performed by an autonomous reasoning agent. The LatchBio whitepaper argues for more end-to-end reasoning across tasks.
So which approach is right?
First off, if you have the resources to keep a human in the loop, it doesn’t really matter which approach you choose, since you can always rest assured that the output has been checked. The AI functions as an assistant that gives the curator leverage on their time.
The more interesting question is: what if you couldn’t have a human verify all the results? Say I wanted to spend a weekend to get high-quality metadata for all scBaseCount datasets, and I only had time to sanity-check a handful of them. Which approach would give me the most accurate answers?
Let’s consider the tradeoffs.
The router’s logic is more set in stone, so maybe it won’t be able to handle an edge cases that doesn’t fit the programmer’s worldview. The agent can reason flexibly, but this means it can also go off the rails.
We’ll only know for sure when the benchmark I proposed gets built.
Conclusion
Metadata curation remains an important problem, even as self-supervised foundation models grab more and more headlines.
AI agents can certainly help, but as of today, they do tasks middle-to-middle, not end-to-end. The two bottlenecks are writing good prompts and verifying the outputs. Routers require more attention to the prompting, whereas agents will (likely) require more attention to verification.
It will be fun to see how companies and academics navigate this idea maze.
Thanks for reading! If you spot any mistakes in this article, please let me know in the comments and I’ll correct them. Also, views are my own and should not be construed as my employer’s.
A clever reader might smirk and say “large corpus of single-cell data, and then what??”. They could argue that early data-slurping foundation model efforts have not panned out. They might argue that the real value lies in perturbational data. They could say that single-cell data is too noisy and you must replace/combine it with a different data modality. This is a fun discussion to have, and I have a lot of thoughts on it that I’ll punt to a future article. For now, suffice to say that this “clean single-cell data corpus” is a product sold by several vendors, so clearly the customers find value in it for the exploratory use case. In addition, it’s also possible to imagine a data cleaning AI agent repurposed for a different data modality.
“Good, fast, or cheap, but you can only pick two” sums up my experience with external vendors. Hopefully an AI agent could be all three.
There is another valid question here: sure, it’s a lot of work, but do you even really need all this metadata? My answer is yes. In the “classical” CompBio case it’s pretty self-explanatory: as an expert human trying to wrap their head around a dataset, you want all the clinical information you can get. For ML use cases, even when the training is fully self-supervised, you still want to validate your model using clinical metadata! For example, NewLimit explains their “looks-like” test to see whether a cell’s transcription profile looks younger. To build a similar old-vs-young classifier, you’d obviously need age as a metadata column.
Sometimes the ontology is overkill and a simple list of allowed terms will do.
If you have ever seen the reasoning trace of a model like Claude 4, it’s funny how similar this self-talk is. I think these end-to-end examples of completed knowledge work tasks, including self-talk, will be considered the new gold for reinforcement learning.
Take a moment to appreciate that since supplementary files are difficult to deal with, the information in them is usually missing in public compendia. This is a big issue! Many research questions can only be answered when this data is available. What differentiates Very Severe Aplastic Anemia from mere Severe Aplastic Anemia? Can we find a cell state that differentiates responders from non-responders? Can the baseline cytogenetics help us inform such a prediction?
If you hire an external data vendor, they’ll typically charge you for around 8 hours. Rarely does it cost less than $1,000 per dataset!
The scope of this article is metadata curation only, and I didn’t focus on processing the actual genomic data. However, at the end of the day, you still need it. As I said, the scBaseCount authors did an amazing job here, even though they only include 10x data on SRA. If they wanted to start tackling the long tail of non-10x, non-SRA datasets, they would benefit from a benchmark that included lots of those examples.
Many datasets don’t live in SRA – they could live on several other sequencing data portals, some of which might be controlled access (like EGA or dbGap). They might also live on generic data repos like Zenodo, Figshare, Dropbox or even an academic lab’s website (see Bassez et al. for an example).
It’s worth noting here that Elucidata’s paper only benchmarked on nice datasets. I would love to see a v2 of the benchmark that includes more challenging examples as well.
For example, they might shift all the table’s rows down by one, thus ruining all annotations.
Structured outputs are very helpful here.